Моногенные патологии это заболевания, вызванные изменением только одного гена.
Ген – это небольшой участок ДНК. Все гены находятся в хромосомах – длинных молекулах, расположенных в клеточном ядре.
Ген – это инструкция для построения организма.
Генетические изменения могут возникать спонтанно или передаваться от родителей.
Виды наследования моногенных заболеваний
В медицине различают несколько видов наследования генетических заболеваний.
Основные виды наследования генетических болезней:
- аутосомные – не зависят от пола ребенка;
- сцепленные с полом ребенка.
Вероятность возникновения генетической мутации у ребенка от родителей зависит от типа наследования болезни – будет ли она аутосомно-доминантной или аутосомно-рецессивной.
При аутосомно-доминантной болезни ребенок унаследует одну нормальную копию гена и одну сломанную от родителей. Вероятность того, что мутация окажется “сильнее” и подавит здоровый ген, составляет 50%.
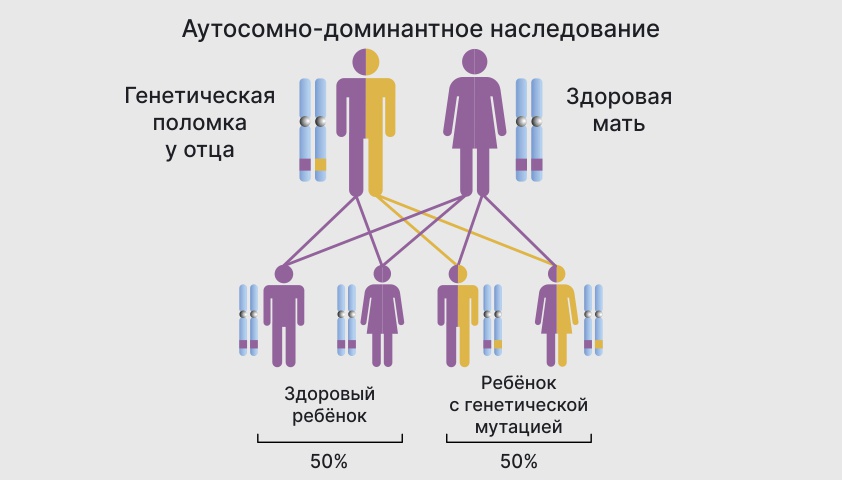
Шанс рождения ребенка с патологией при аутосомно-доминантном наследовании – 50%
В случае аутосомно-рецессивного заболевания ребенок получает по одной копии сломанного гена от каждого родителя – носителя мутации. У матери и отца признаков болезни может и не быть, но у ребенка болезнь проявится.
Шанс унаследовать рецессивное заболевание составляет 25%. В 50% случаев дети, пораженные этими мутациями, становятся только переносчиками. В 25% случаев генетической мутации не произойдет.
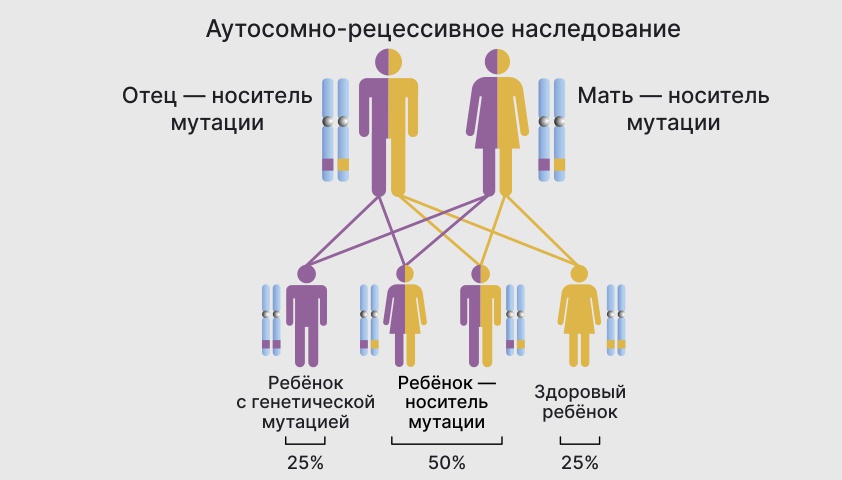
Вероятность появления ребенка с патологией при аутосомно-рецессивном наследовании составляет 25%
Муковисцидоз и спинальная мышечная атрофия (СМА) относятся к числу наиболее распространенных аутосомно-рецессивных заболеваний.
Генетические заболевания, связанные с полом, возникают, когда у ребенка возникает мутация в одной из половых хромосом — X или Y.
Исследователи выяснили, что один из генов X-хромосомы отвечает за цветовое зрение. В том случае, если от матери мальчик получает копию такого гена с мутацией, у него развивается дальтонизм: в большинстве случаев ребенок различает только желтый и синий цвета, в то время как красный и зеленый он воспринимает как похожие.
Тест Ишихары помогает выявить дальтонизм. Люди с таким особенным зрением не способны увидеть цифры 8, 9, 12, 7 на этом изображении
Носительство моногенных заболеваний
У носителей моногенных заболеваний некорректно функционирует только одна копия гена. Вторая копия компенсирует дефект, и болезнь не проявляется.
Однако опасность носительства заключается в том, что человек может передать дефектный ген своим потомкам, что может привести к проявлению болезни у них.
Если оба родителя стали носителями генетической мутации, то шанс на появление у них больного ребенка значительно выше.
Данные о моногенных заболеваниях
Частота моногенных заболеваний варьирует от 1 случая на 2 0000 новорожденных (муковисцидоз) до 1 случая на 350 000 (анемия Фанкони).
Опасность этих заболеваний заключается в том, что ребенок может унаследовать их от внешне здоровых родителей – носителей патологии. При этом болезни протекают очень тяжело, а лекарства от них до сих пор не разработаны.
Распространенные моногенные заболевания
Известно около 5000 моногенных заболеваний, и примерно половина из них наследуется по аутосомно-доминантному типу.
Наследственная нейросенсорная тугоухость – одно из самых распространенных моногенных заболеваний, оно обнаруживается у одного ребенка из 1000 новорожденных.
Причиной заболевания являются изменения в одном из следующих генов: SLC26A4, GJB2, LOXHD1, OTOGL, TMPRSS3, MYO6.
Данное заболевание, как правило, проявляется в ранние месяцы жизни малыша. Основные признаки – нарушение слуха и вторичные проблемы с речью.
Муковисцидоз (кистозный фиброз) – частое генетическое заболевание (1 случай на 2 000–2 500 новорожденных), вызванное генетическими изменениями в гене CFTR.
При муковисцидозе нарушается функционирование желез внешней секреции, таких как бронхиальные железы, поджелудочная железа, печень, потовые железы. Симптомы заболевания связаны с тем, что секрет этих желез становится вязким и густым, что затрудняет его выделение. В результате у пациентов могут развиться тяжелые осложнения, такие как тяжелая пневмония, дыхательная и сердечная недостаточность, сахарный диабет, цирроз печени, легочные и желудочные кровотечения и другие проблемы.
Генетические формы синдрома Ушера: тип 1С, 1D, 1B, 2A, 3A — это редкие заболевания, которые диагностируются лишь у одного новорождённого из 6 000. Причиной этой болезни становятся изменения в генах, таких как USH2A, CLRN1, MYO7A, CDH23, USH1C, которые нарушают функцию зрительных и слуховых рецепторов. В итоге у малыша развивается угнетение слуха, снижение зрения, а затем, в большинстве случаев, происходит слепоглухота.
Фенилкетонурия — это редкое генетическое заболевание (встречается лишь у 1 из 10 000 новорождённых), вызванное мутацией в гене PAH. Дети, страдающие этим состоянием, врожденно кажутся здоровыми. Но они не могут обработать фенилаланин — аминокислоту, содержащуюся в грудном молоке, молочной смеси и других белковых продуктах. Со временем фенилаланин накапливается в организме и вредит нервную систему.
Ранние признаки фенилкетонурии: тошнота, аромат плесени из мочи и кожи, замедление психомоторного развития. Поздние: проблемы с физическим развитием, судороги, экзема на коже.
Синдром Ретта – заболевание, которое встречается у одной из каждых 10 000-15 000 новорожденных девочек (у мальчиков это редко случается). Из-за мутаций в гене MECP2 нарушается работа нервной системы, прогрессирует умственная отсталость, мышцы ослабевают. Дети, страдающие этим заболеванием, не могут контролировать свои движения, а их позвоночник деформируется.
Врожденные нарушения гликозилирования типов 1A, 1B, 1C, 1K, 1P – группа генетически обусловленных заболеваний, вызванных мутациями в одном из генов: MPI, PMM2, ALG1, ATP7B, ALDOB. По статистике, такие патологии встречаются у одного ребенка из 10 000 новорожденных.
Гликозилирование – это процесс, при котором к белкам организма при участии ферментов присоединяются новые «строительные блоки» (остатки сахаров) и образуются гликопротеины. Этот процесс очень важен для нормального развития детей.
При нарушении гликозилирования возникает задержка в физическом и умственном развитии, нарушается свертываемость крови, появляются патологии сердца, печени.
Аутосомно-рецессивная поликистозная болезнь почек – наследственное заболевание, которое обнаруживается у одного новорожденного из 20 000. Причина болезни – мутации в гене PKHD1.
Вследствие генетического дефекта на поверхности и внутри почек образуются различные кисты. Постепенно они увеличиваются и замещают здоровую ткань. В результате функция почек по фильтрации крови и выведению мочи нарушается. Развивается хроническая почечная недостаточность.
Мукополисахаридозы типов 1H, 2, 3A, 3C – наследственные моногенные заболевания, вызванные мутациями в одном из генов: IDUA, IDS, HGSNAT, SGSH.
У пациентов с такими нарушениями нарушается обмен веществ, не хватает ферментов, способных расщеплять длинные молекулы сахара – мукополисахариды. В результате сахар накапливается в тканях и органах, постепенно разрушая их. Основные симптомы болезни: задержка физического развития, умственная отсталость, изменения в костях и хрящах, грыжи, увеличение печени и селезенки, проблемы с сердцем.
Наследственная фруктозурия (непереносимость фруктозы) – редкое генетическое заболевание, которое встречается у одного ребёнка из 23 000-40 000 человек. Из-за изменения в гене ALDOB нарушается обмен фруктозы – фруктового сахара, содержащегося в мёде, яблоках, грушах и черносливе. В результате продукты обмена этого вещества накапливаются в организме и разрушают различные органы и системы.

Людям с наследственной фруктозурией необходимо исключить из рациона продукты, содержащие фруктозу.
Если пациент с таким диагнозом употребит продукт, содержащий фруктозу, у него могут появиться тошнота, рвота и боль в животе. Без коррекции рациона со временем могут возникнуть почечная и печеночная недостаточность.
Заболевание Баттена, известное также как нейрональный цероидный липофусциноз, — генетическое заболевание, вызванное изменением одного из генов: TPP1, PPT1, CLN6. Частота появления этого заболевания составляет 1 случай на 25 000.
Люди, страдающие от болезни Баттена, накапливают в организме пигмент липофусцин, который является токсичным для клеток и приводит к их разрушению. Симптомы этого заболевания включают приступы эпилепсии, потерю зрения, прогрессирующие нарушения двигательных функций и умственные расстройства.
Синдром Смита — Лемли — Опица — нарушение обмена холестерина, вызванное изменениями в гене DHCR7. Это заболевание обнаруживается у одного новорожденного из 29 000.

Мутация гена может привести к дефектам развития у ребенка, таким как наличие дополнительных пальцев на руках (полидактилия)
Основные признаки данного генетического заболевания: низкий рост, умственная отсталость, деформация пальцев рук и ног, дефекты развития внутренних органов.
Гепатоцеребральная дистрофия Вильсона — Вестфаля — Коновалова (болезнь Вильсона) — генетическое заболевание, вызванное изменениями в гене ATP7B. Встречаемость составляет 1 случай на 30 000–100 000 новорожденных.
У пациентов с данным заболеванием медь не выводится из организма, а накапливается во внутренних органах, постепенно их разрушая. Люди, страдающие болезнью Вильсона — Вестфаля — Коновалова, часто испытывают проблемы с сердцем и печенью, нарушения свертываемости крови, неврологические нарушения.
Галактоземия — это патология, связанная с изменениями в гене GALT. Генетические нарушения приводят к тому, что организм не может усваивать галактозу, содержащуюся в большинстве молочных продуктов.
У детей с галактоземией симптомы начинают проявляться с первых дней жизни: желтуха, увеличение печени, нистагм (непроизвольные движения глаз), судороги, рвота. По мере прогрессирования болезни появляется катаракта. Ребенок отстает в физическом и умственном развитии.
Частота встречаемости составляет 1 случай на 40 000–60 000 новорожденных.
Нейрофиброматоз II типа — это редкое (1:50 000) генетическое заболевание, связанное с изменениями в гене NF2.
Больные этим заболеванием имеют нарушения работы белка нейрофибромина-2, который должен подавлять рост опухолей. Из-за неполадок в работе белка возникают множественные доброкачественные опухоли в центральной нервной системе и вдоль периферических нервов.
Тирозинемия I типа – крайне редкое заболевание (1 случай на 100 000–120 000 новорожденных), которое проявляется у людей с мутациями в гене FAN. У пациентов с этим диагнозом недостаточно фермента, участвующего в метаболизме аминокислоты тирозина. Это приводит к накоплению токсичных веществ в крови, которые негативно воздействуют на печень и нарушают свертываемость крови. У детей с тирозинемией часто наблюдается задержка в умственном и физическом развитии.
Заболевание Андерсона-Фабри – наследственное заболевание, вызванное дефектом гена GLA. Это распространенная патология, встречающаяся в 1 случае на 117 000 новорожденных.
При заболевании Андерсона-Фабри происходит недостаток фермента α-галактозидазы A. В органах накапливаются жировые метаболиты, что приводит к поражению в первую очередь почек, сердца и сосудов мозга.
Болезнь Тея-Сакса – очень редкое заболевание, которое встречается у одного из 120 000 новорожденных, а у евреев ашкенази этот показатель намного выше – 1 случай на 3 000 человек.

Ашкенази – этническая группа евреев, проживающая в Центральной и Восточной Европе.
Мутации в гене HEXA приводят к накоплению фрагментов клеточных оболочек в нейронах, что приводит к их разрушению. Этот процесс негативно сказывается на головном и спинном мозге.
Симптомы болезни Тея-Сакса включают трудности с глотанием, нарушение фокусировки взгляда, проблемы с двигательной активностью, снижение зрения и слуха, задержка умственного развития, а также судороги.
Анемия Фанкони — генетическое заболевание, вызванное мутациями в генах FANCA, FANCC, FANG. Его частота составляет 1 случай на 350 000 новорожденных.
У пациентов с анемией Фанкони нарушается образование крови, возникают злокачественные опухоли и дефекты развития. Симптомы болезни включают частые кровотечения, синяки на коже, вялость, бледность и повышенную восприимчивость к инфекциям.
Методы диагностики генетических заболеваний

Идентификация мутаций в генах и оценка вероятности рождения ребенка с моногенным заболеванием осуществляются с помощью генетического исследования.
Отследить мутации в одном из генов, вызывающих наследственную нейросенсорную тугоухость (SLC26A4, GJB2, LOXHD1, OTOGL, TMPRSS3, MYO6), и определить или подтвердить данный диагноз помогает точный анализ.
Диагностирование муковисцидоза осуществляется специальными генетическими исследованиями.
Синдром Ушера типов 1C, 1D, 1B, 2A, 3A – серьезное моногенное заболевание, которое чаще всего приводит к слепоте и глухоте ребенка, – можно выявить с помощью генетического теста.
Определение мутаций в генах, способных привести к фенилкетонурии, выполняется генетическим тестированием.
Определиться с синдромами Ретта, синдромом Смита-Лемли-Опица, а также болезнью Вильсона-Вестфаля-Коновалова, болезнью Баттена, болезнью Тея-Сакса и болезнью Андерсона-Фабри помогают точные генетические исследования, которые можно провести в Лаборатории Гемотест.
Также генетические тесты помогают выявить наследственную непереносимость фруктозы (фруктозы), что позволяет своевременно разработать специальный рацион.
В Лаборатории Гемотест можно сдать анализы на самые редкие генетические патологии, такие как аутосомно-рецессивная болезнь почек, мукополисахаридоз, нейрофиброматоз II типа и другие.
Кроме того, всем, кто планирует беременность, желает стать донором спермы или яйцеклеток, рекомендуется пройти комплексные исследования на наличие наиболее распространенных моногенных заболеваний.
Какие действия необходимо предпринять носителю после диагностики
У носителя генетического заболевания мутация присутствует только в одной копии гена, и поэтому, как правило, этот человек остается здоровым и не имеет симптомов болезни, поскольку неповрежденная копия гена компенсирует дефект.
По статистике, большинство людей – носители различных генетических заболеваний.
Однако, если оба партнера, планирующие ребенка, имеют одинаковую мутацию, то у малыша с высокой вероятностью начнет проявляться эта болезнь. Проведение генетических исследований позволяет заранее оценить риск рождения больного ребенка.
Источники

- Бочков Н. П. Клиническая генетика. М., 2001.
- Thomson G., Esposito M. S. The genetics of complex diseases // Trends Cell Biol. 1999. Vol. 9(12). P. M17—20.
Генетическое консультирование для носителей
Генетическое консультирование включает в себя обширный анализ и оценку генетической информации, собранной о пациенте и его семье. Специалисты в этой области помогают пациентам понять риск передачи генетических заболеваний и принимают участие в разработке индивидуальных стратегий профилактики и лечения.
- Консультации проводятся генетиками и генетическими консультантами, которые обладают специализированными знаниями в области генетики и медицины.
- Процесс включает в себя сбор семейного анамнеза, проведение генетических тестов, интерпретацию результатов и разработку рекомендаций по дальнейшему наблюдению и лечению.
- Целью генетического консультирования является не только предупреждение наследственных заболеваний, но и обеспечение пациентов информацией и поддержкой в принятии важных жизненных решений.
Генетическое консультирование для носителей играет ключевую роль в поддержании здоровья семей и предотвращении возможных генетических рисков для будущих поколений. Пациенты, получающие такие консультации, имеют возможность принимать осознанные решения, основанные на научных данных и рекомендациях экспертов в области генетики.
Генетическое консультирование для носителей
| Генетическое тестирование | Проведение тестов для выявления наличия генов, ответственных за наследственные заболевания. Позволяет определить риск передачи генов потомкам. |
| Индивидуальное консультирование | Получение персональных рекомендаций и советов по предотвращению передачи генов наследственных заболеваний. Учитываются индивидуальные особенности и риски. |
| Психологическая поддержка | Помощь психолога в решении проблем, связанных с наследственными заболеваниями и их передачей. Предоставляются советы по преодолению стресса и адаптации. |
Генетическое консультирование для носителей генов наследственных заболеваний играет важную роль в предупреждении и преодолении возможных рисков передачи таких заболеваний. Оно позволяет людям принимать информированные решения относительно их здоровья и здоровья их потомков, а также обеспечивает необходимую поддержку и сопровождение в этом процессе.
Этические аспекты генетической диагностики
В данном разделе рассматриваются важные вопросы, связанные с этикой при проведении генетической диагностики. Основное внимание уделяется нравственным дилеммам, возникающим при анализе генетической информации и принятии соответствующих решений.
| Одним из главных аспектов обсуждения является приватность и конфиденциальность генетических данных пациента. Важно обеспечить защиту личной информации, предотвращая ее неправомерное раскрытие и использование. |
| Другим важным вопросом является согласие на генетическое тестирование. Пациент должен быть в полном понимании последствий проведения анализа и иметь возможность принимать решение об участии в нем. |
| Также стоит обсудить этические аспекты связанные с тем, как используются полученные данные. Необходимо учитывать интересы пациента и обязательство сохранять его достоинство, уважая его собственные предпочтения и жизненные ценности. |
Исследования рода и профилактика заболеваний
Родоведческие исследования играют важную роль в предупреждении наследственных заболеваний и сохранении здоровья будущих поколений. Путем анализа генетической информации предков можно выявить потенциальные риски для заболеваний и принять меры по их предотвращению.
Специалисты проводят обширные генетические исследования для выявления возможных наследственных патологий в роду. Это помогает определить группы риска и принять меры по профилактике заболеваний, в том числе разработать индивидуальные программы мониторинга и лечения.
Исследования рода позволяют также выявить носителей генетических мутаций и предложить им генетическое консультирование. Это позволяет снизить риск передачи наследственных заболеваний потомкам и принять меры по их предотвращению в будущем.
Видео по теме:
Вопрос-ответ:
Чем отличаются моногенные заболевания от многофакторных?
Моногенные заболевания вызваны мутацией только одного конкретного гена, в то время как многофакторные заболевания обусловлены воздействием нескольких генов и внешних факторов.
Как можно диагностировать моногенные заболевания?
Диагностика моногенных заболеваний чаще всего проводится с помощью генетических тестов, которые позволяют выявить наличие или отсутствие мутации в конкретном гене.
Могут ли моногенные заболевания быть унаследованы от родителей?
Да, моногенные заболевания могут быть унаследованы по наследственной линии от одного или обоих родителей, если у них также есть мутация в том же гене.
Какие могут быть последствия моногенных заболеваний для организма?
Последствия моногенных заболеваний могут быть разнообразными, включая нарушения в развитии тканей и органов, серьезные заболевания и иногда даже смертельные исходы, в зависимости от конкретного типа мутации и пораженного гена.